将二氧化碳转化为甲烷不仅为可再生能源提供了前景广阔的替代方案,更在减少碳排放方面发挥着关键作用。然而,二氧化碳高效转化为甲烷仍面临重大挑战,包括反应动力学、催化剂失活、温度控制和平衡限制等问题,亟需开发先进的催化系统以提升反应效率,推动工业化应用。
镍(Ni)在二氧化碳甲烷化反应中展现出优异的经济性和催化性能,是贵金属催化剂的一种极具成本效益的替代选择,但其需要超过350℃的高温才能有效运行,且易烧结导致失活。因此,设计能在低温下高效运行的镍基催化剂对二氧化碳氢化反应至关重要。

近期,清华大学化工系王玉军教授课题组与中国科学技术大学王占东教授课题组合作,通过微流控合成技术制备Ni/CeO2催化剂,显著提升了其在低温CO2甲烷化反应中的性能,揭示了氧空位和金属分散度对催化效率的关键影响。相关研究以 “Tailoring Ni/CeO2 oxygen vacancies and metal dispersion via microfluidic synthesis techniques for enhanced low-temperature CO2 methanation” 为题目,发表于期刊《Chemical Engineering Journal》。
本文要点:
1、本研究采用膜分散(MD)、小直径T型结(STJ)和T型结(TJ)微反应器三种连续合成技术制备Ni/CeO2催化剂,并与简单共沉淀(SC)法对比。
2、结果表明,250℃、空速30,000 ml・g-1・h-1条件下,STJ、MD、TJ的CO2转化率分别为80%、77%、72%,远超SC的9.14%,且CH4选择性均为100%。
3、MD催化剂在300℃时表现出175 mmol・g-1・h-1的高空时产率,80h内无失活,CO的形成可以忽略不计。
4、原位表征和DFT计算证实,微流控技术可提高Ni分散度和氧空位浓度,强化Ni-CeO2相互作用,加速甲酸盐路径中氢化物物种的生成,并降低反应吉布斯能。
微流控合成法(包括膜分散(MD)、小直径T型结(STJ)、T型结(TJ)微反应器法)与传统共沉淀法(SC)在制备Ni/CeO₂催化剂上的区别主要体现在以下方面:
1、合成过程:传统共沉淀法是在烧杯中混合金属溶液与沉淀剂溶液,以600rpm搅拌6小时;微流控合成法通过微反应器进行连续合成,总反应时间仅75s,其中MD法利用恒流泵将分散相和连续相送入膜分散微反应器,产物经PTFE延时管进行水热处理,TJ和STJ法也遵循类似连续流程。
2、催化性能:250℃、GHSV为30,000 ml・gcat⁻¹・h⁻¹时,STJ、MD、TJ的CO₂转化率分别约为80%、77%、72%,远高于SC的9.14%,且均保持100% CH₄选择性;MD在300℃时甲烷时空产率达175 mmol・gcat⁻¹・h⁻¹,80小时无失活,而SC性能较差且CO产率较高。
3、结构特性:微流控合成的催化剂Ni分散度更高(MD为9.65%、STJ为8.83%、TJ为8.42%,SC为7.46%),Ni颗粒尺寸更小(MD约6.13nm、STJ约6.12nm,SC约9.86nm),氧空位浓度更高(ID/IF2g比值MD为0.88、STJ为0.84,SC为0.29),且Ni-CeO₂相互作用更强。
4、反应相关特性:微流控合成的催化剂(尤其是MD和STJ)H₂消耗更多,具有更丰富的中等强度CO₂-TPD活性位点,通过甲酸盐路径生成甲烷的速率更快,而SC在这些方面表现较弱。
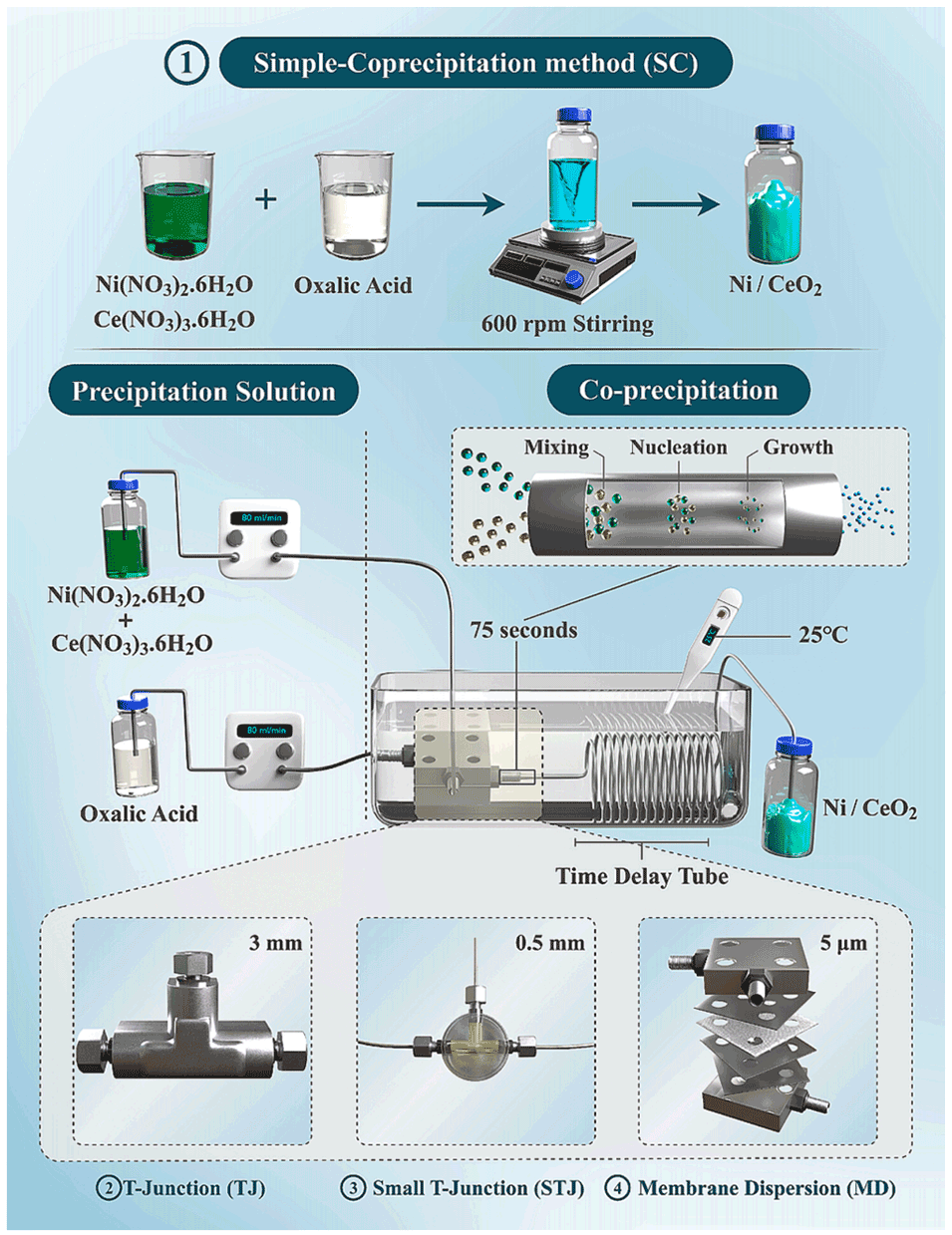
Scheme1:简单共沉淀法(SC)、T型结(TJ)、小直径T型结(STJ)及膜分散(MD)微反应器系统的连续合成方案示意图。
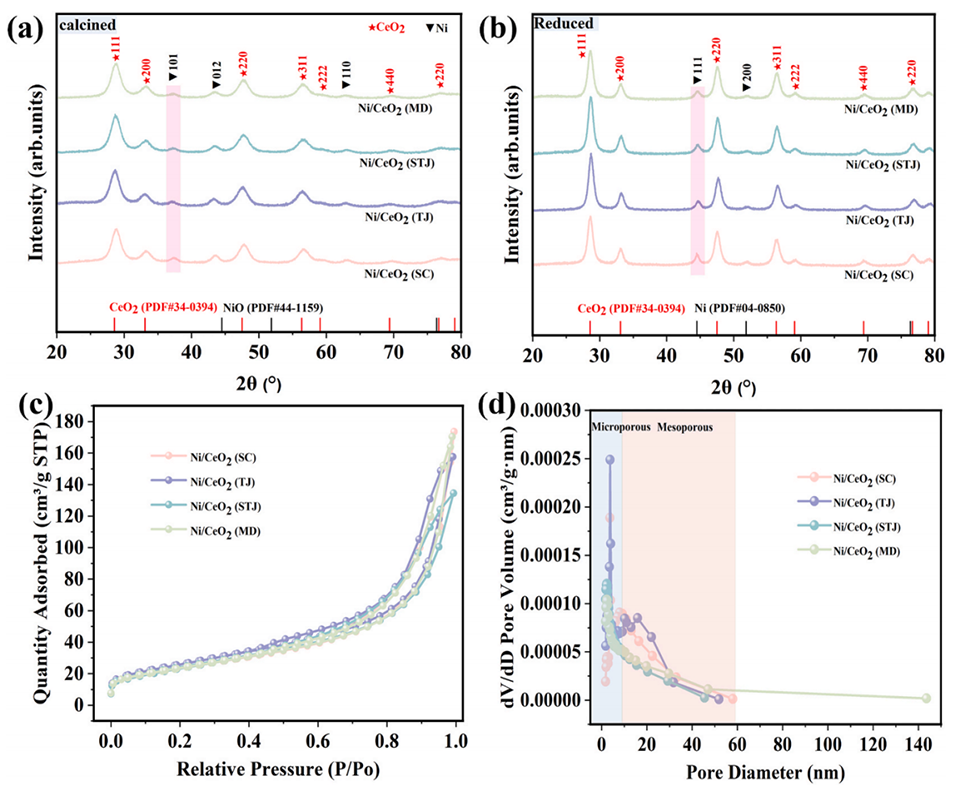
图1 :(a)煅烧态、(b)还原态Ni/CeO2(SC)、Ni/CeO2(TJ)、Ni/CeO2(STJ)和Ni/CeO2(MD)催化剂的XRD图谱;(c)N2吸附/脱附曲线;(d)孔径分布曲线。
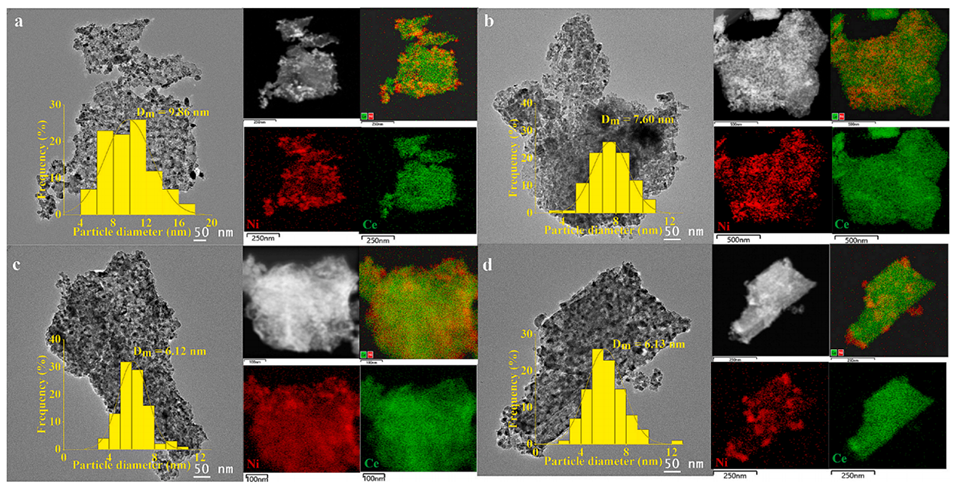
图2:(a)Ni/CeO2(SC)、(b)Ni/CeO2(TJ)、(c)Ni/CeO2(STJ)和(d)Ni/CeO2(MD)催化剂的高分辨透射电镜图像、镍颗粒尺寸分布直方图及能谱分析图。
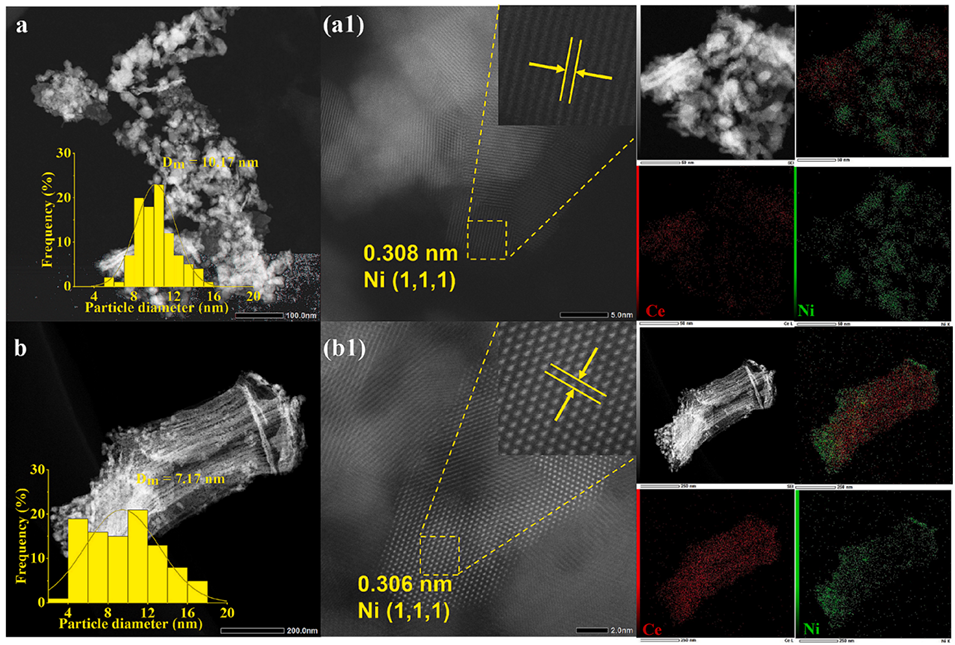
图3:(a)Ni/CeO2(SC)和Ni/CeO2(MD)还原态催化剂的高角度环形暗场扫描透射电镜图像、镍颗粒尺寸分布直方图及能谱分析图。
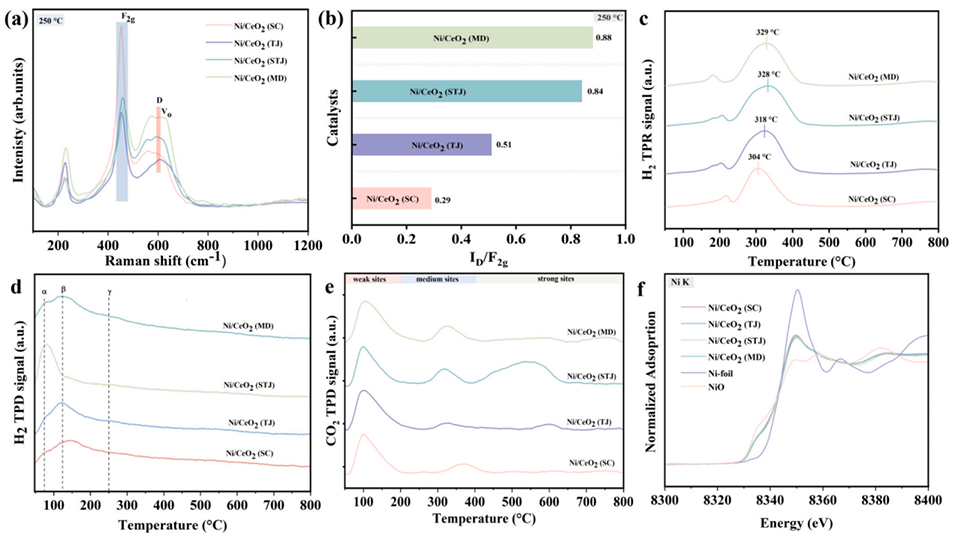
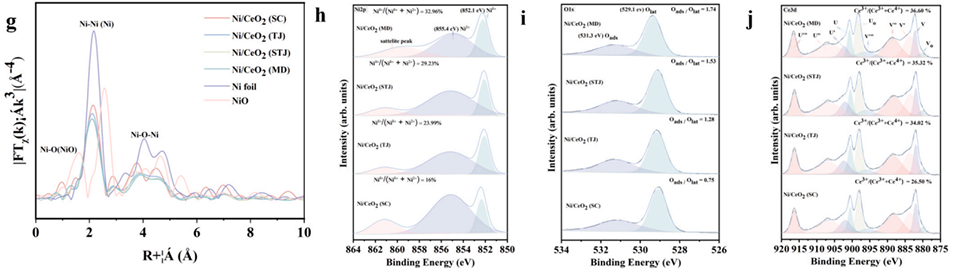
图4:(a)制备的催化剂在250℃下的原位拉曼光谱峰对比;(b)250℃下ID/IF2g比值对比;(c)H2程序升温还原;(d)H2程序升温脱附;(e)CO2程序升温脱附分析。还原态Ni/CeO2催化剂(镍箔、氧化镍)的X射线吸收精细结构表征光谱;(f)Ni K边X射线吸收近边结构光谱;(g)Ni K边扩展X射线吸收精细结构光谱。Ni/CeO2(SC)、Ni/CeO2(TJ)、Ni/CeO2(STJ)、Ni/CeO2(MD)催化剂的准原位X射线光电子能谱:(h)Ni2p;(i)O1s;(j)Ce3d。
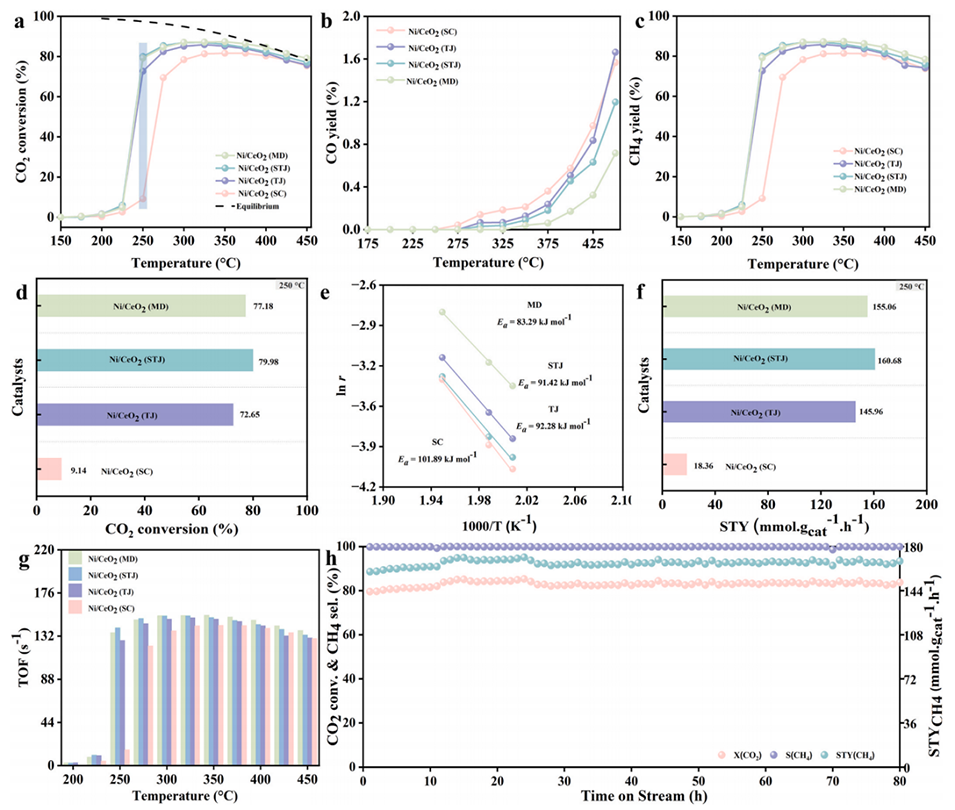
图5:(a)Ni/CeO2(SC、TJ、STJ、MD)催化剂的CO2转化率,(b)CO产率,(c)CH4产率。(d)250℃下CO2转化率对比;(e)Ni/CeO2(SC、TJ、STJ、MD)催化剂的阿累尼乌斯图;(f)250℃下所有催化剂的甲烷时空产率对比;(g)所有制备催化剂的转换频率;(h)Ni/CeO2(MD)催化剂在300℃下的长期稳定性测试。反应条件:100mg(纯催化剂粉末),混合气体(CO2: H2: N2=1:4:1.5),流速50mL/min,1atm,体积空速=30000ml・gcat-1・h-1。
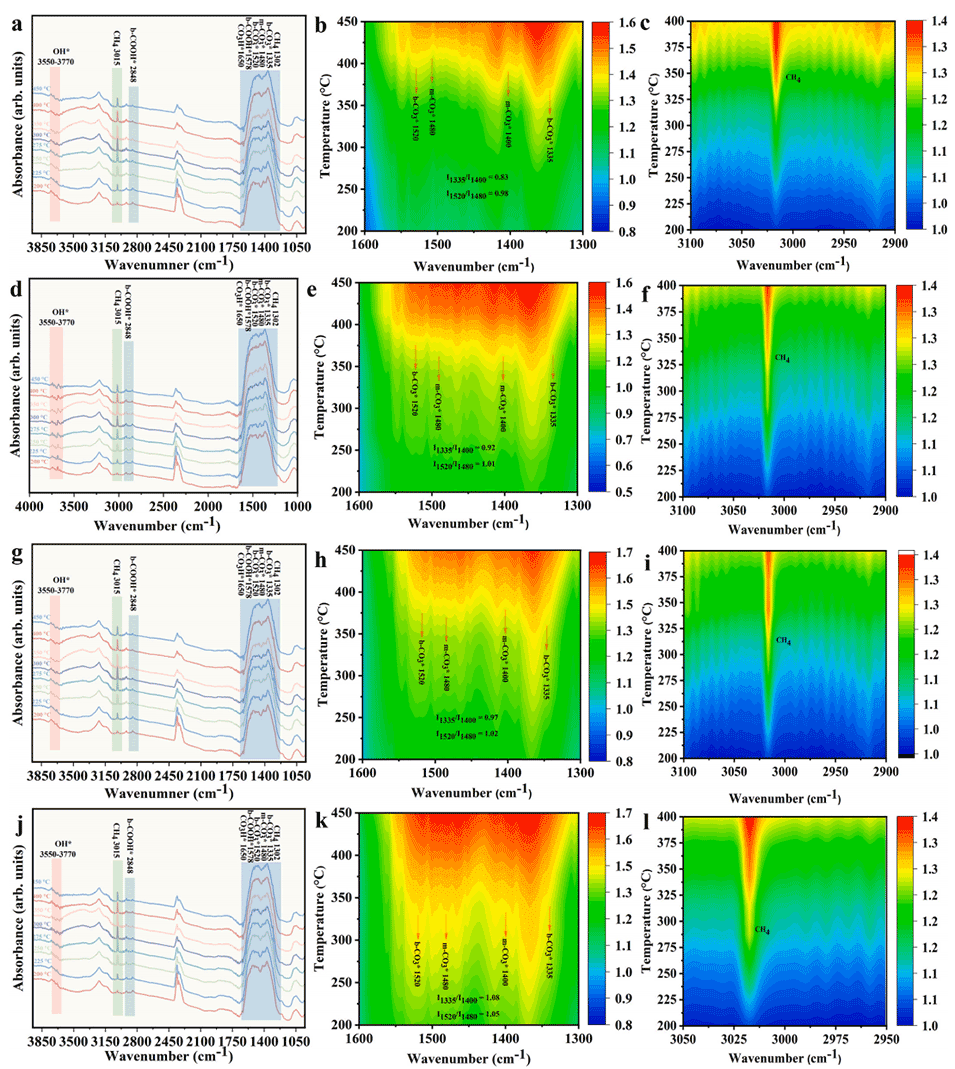
图6:将H2切换为H2+CO2 30分钟后,(a-c)Ni/CeO2(SC)、(d-f)Ni/CeO2(TJ)、(g-i)Ni/CeO2(STJ)和(j-l)Ni/CeO2(MD)催化剂随反应温度变化的原位漫反射红外傅里叶变换光谱。
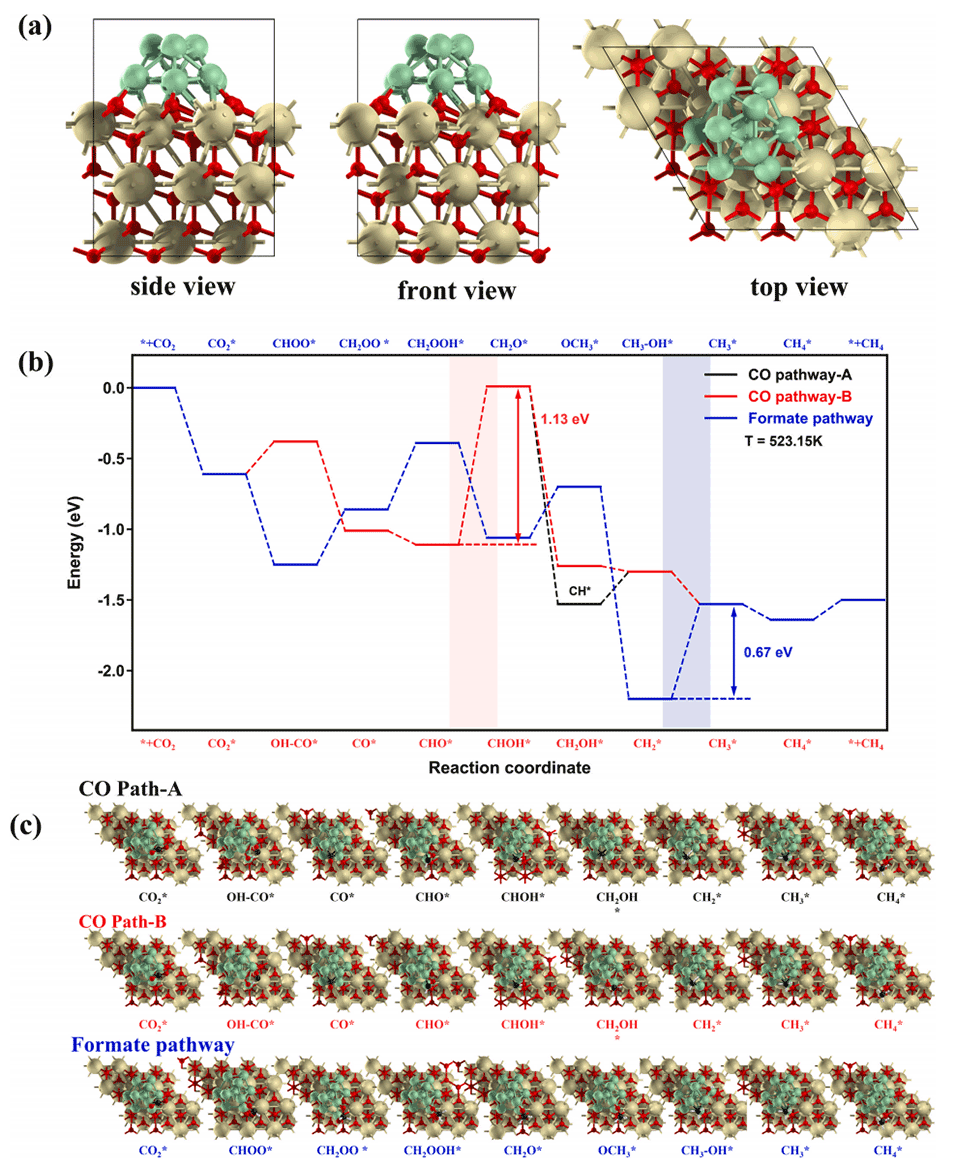
图7:(a)Ni10团簇在CeO2(111)表面吸附的侧视图、正视图和顶视图;(b)250℃下不同路径的吉布斯自由能曲线;(c)CO2甲烷化反应中不同Ni/CeO2中间体最稳定的吸附构型。
论文链接:https://doi.org/10.1016/j.cej.2025.163859